Cell Correction¶
This part shows how to correct cells in Stereopy. Optionally, there are two kinds of input to complete it:
correcting from BGEF and mask;
correcting from GEM and mask.
Provided with three algorithmic methods, you could determine which one to perform by setting the parameter method.
method=GMMis based on GMM (Gaussian Mixture Model) algorithm, which performs cell correction using both gene expression matrix and spatial information, with much time and memory consumption. Multi-processing would be used if setprocess_countto more than 1.method=FASTperforms correction based on the distance between spot and centroid of the cell, when the distance is less than adjusting threshold, the spot is considered to belong to the cell. It only supports single process and single threading.method=EDMis based on EDM (Euclidean Distance Map) algorithm, which performs correction using mask image, out of cell segmentation. We highly recommnend EDM method that is used by default. Multithreading would be on if setprocess_countto more than 1.
More details refer to API.
Attention
In order to interpret the algorithm of cell correction clearly, the parameter of which method to use has been changed. But nothing to do with the algorithms themselves.
fast=False→method='GMM'fast='v1'→method='FAST'fast='v2'→method='EDM'
Note
The images used here must have been calibrated.
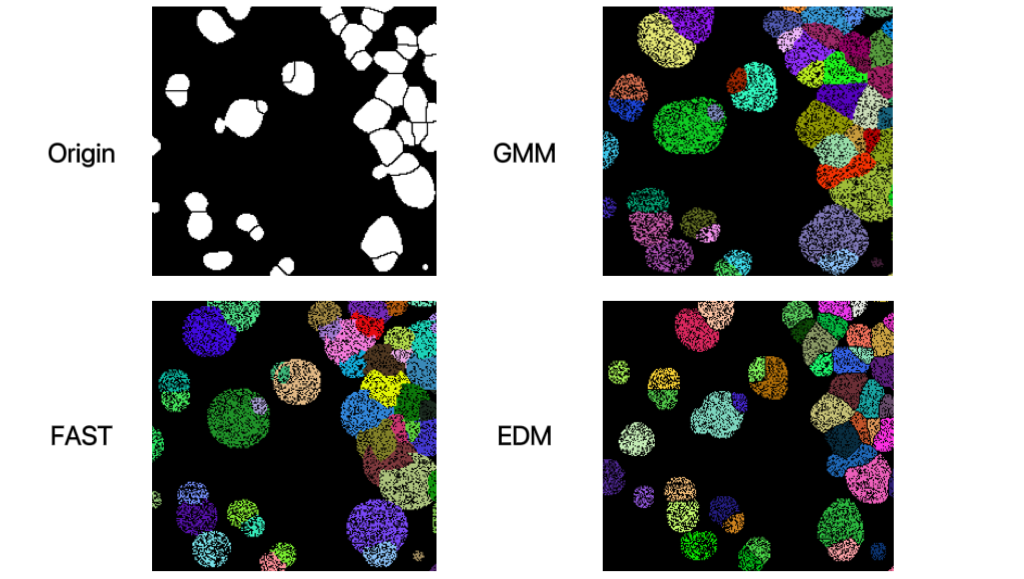
EDM algorithm is expected to eliminate the overlapping influence as much as possible, you can obvisouly learn from the following image that there is almost no overlap after cell correcting.
Correcting from BGEF and mask¶
On this way, you should specify the path of BGEF by bgef_path, the path of mask by mask_path and the output path to save corrected result by out_dir.
Cell correction dafaults to return a StereoExpData object, if set only_save_result to True, only return the path of CGEF after correcting.
If you have no mask currently, you can generate it from ssDNA image, refer to Cell Segmentation.
[ ]:
from stereo.tools.cell_correct import cell_correct
bgef_path = "SS200000135TL_D1.raw.gef"
mask_path = "SS200000135TL_D1_mask.tif"
out_dir = "cell_correct_result"
data = cell_correct(
out_dir=out_dir,
bgef_path=bgef_path,
mask_path=mask_path,
only_save_result=False,
fast='EDM'
)
Output directory includes such files:
.raw.cellbin.gef- the CGEF without correcting, generated from BGEF and mask;.adjusted.gem- the gem after correction;.adjusted.cellbin.gef- the CGEF after correcting, generated from the.adjusted.gem;err.log- records the cells which cannot be corrected and not contained in.adjusted.gemand.adjusted.cellbin.gef.
Correcting from GEM and mask¶
In this way, you should also specify the path of BGEF by bgef_path, the path of mask by mask_path and the output path to save corrected result by out_dir.
In output directory, the file named *.bgef is generated form mask.
[ ]:
from stereo.tools.cell_correct import cell_correct
gem_path = "SS200000135TL_D1.cellbin.gem"
mask_path = "SS200000135TL_D1_mask.tif"
out_dir = "cell_correct_result"
data = cell_correct(
out_dir=out_dir,
gem_path=gem_path,
mask_path=mask_path,
only_save_result=False,
fast='EDM'
)
Running on Jupyter Notebook¶
Notebook can not support multiprocess directly, we recommend following two steps to improve performance.
Firstly, write the source code in a python file by command %%writefile.
[ ]:
%%writefile temp.py
from stereo.tools.cell_correct import cell_correct
bgef_path = "SS200000135TL_D1.raw.gef"
mask_path = "SS200000135TL_D1_mask.tif"
out_dir = "cell_correct_result"
data = cell_correct(
out_dir=out_dir,
bgef_path=bgef_path,
mask_path=mask_path,
process_count=10,
only_save_result=False,
fast='GMM'
)
Secondly, run the file by command %run:
[ ]:
%run temp.py
Note
We strongly suggest you output in CGEF format for subsequent analysis. When you transform CGEF to CGEM for intuitive understantding, there will be a small quantity of genes which are lost because of the algorithm, related to cell boarder. If you are concerned about the lost, just make a comparasion on the gene expression amount of the missing part.
Performance¶
Take a GEF which contains 56204 cells and 24661 genes as an example.
Machine configuration as below:
physical cores |
logic cores |
memory |
|---|---|---|
12 |
48 |
250G |
Comparision of performance:
fast='GMM'
process |
memory(max) |
cpu |
time |
|---|---|---|---|
10 |
120G |
2330% |
2h38m |
fast='FAST' (only support single process)
process |
memory(max) |
cpu |
time |
|---|---|---|---|
1 |
41G |
100% |
26m |
fast='EDM' (single process)
process |
memory(max) |
cpu |
time |
|---|---|---|---|
1 |
15G |
100% |
9m |