


Stereopy - Spatial Transcriptomics Analysis in Python¶
Stereopy is a fundamental and comprehensive tool for mining and visualization based on spatial transcriptomics data, such as Stereo-seq (spatial enhanced resolution omics sequencing) data. More analysis will be added here, either from other popular tools or developed by ourselves, to meet diverse requirements. Meanwhile, we are still working on the improvement of performance and calculation efficiency.
Get quickly started by browsing Usage Principles, Tutorials or API.
Open to discuss and provide feedback on Github.
Follow changes in Release Notes.
News¶
The paper of Stereopy has been pre-printed on bioRxiv!
Upcoming functions¶
Batch Effect removal funciton
Lasso expression matrix and image simultaneously
…
Highlights¶
More suitable for performing downstream analysis of Stereo-seq data.
Support efficient reading and writing (IO), pre-processing, and standardization of multiple spatial transcriptomics data formats.
Self-developed Gaussian smoothing model, tissue and cell segmentation algorithm models, and cell correction algorithm.
Integrate various functions of dimensionality reduction, spatiotemporal clustering, cell clustering, spatial expression pattern analysis, etc.
Develop interactive visualization functions based on features of Stereo-seq workflow.
Workflow¶
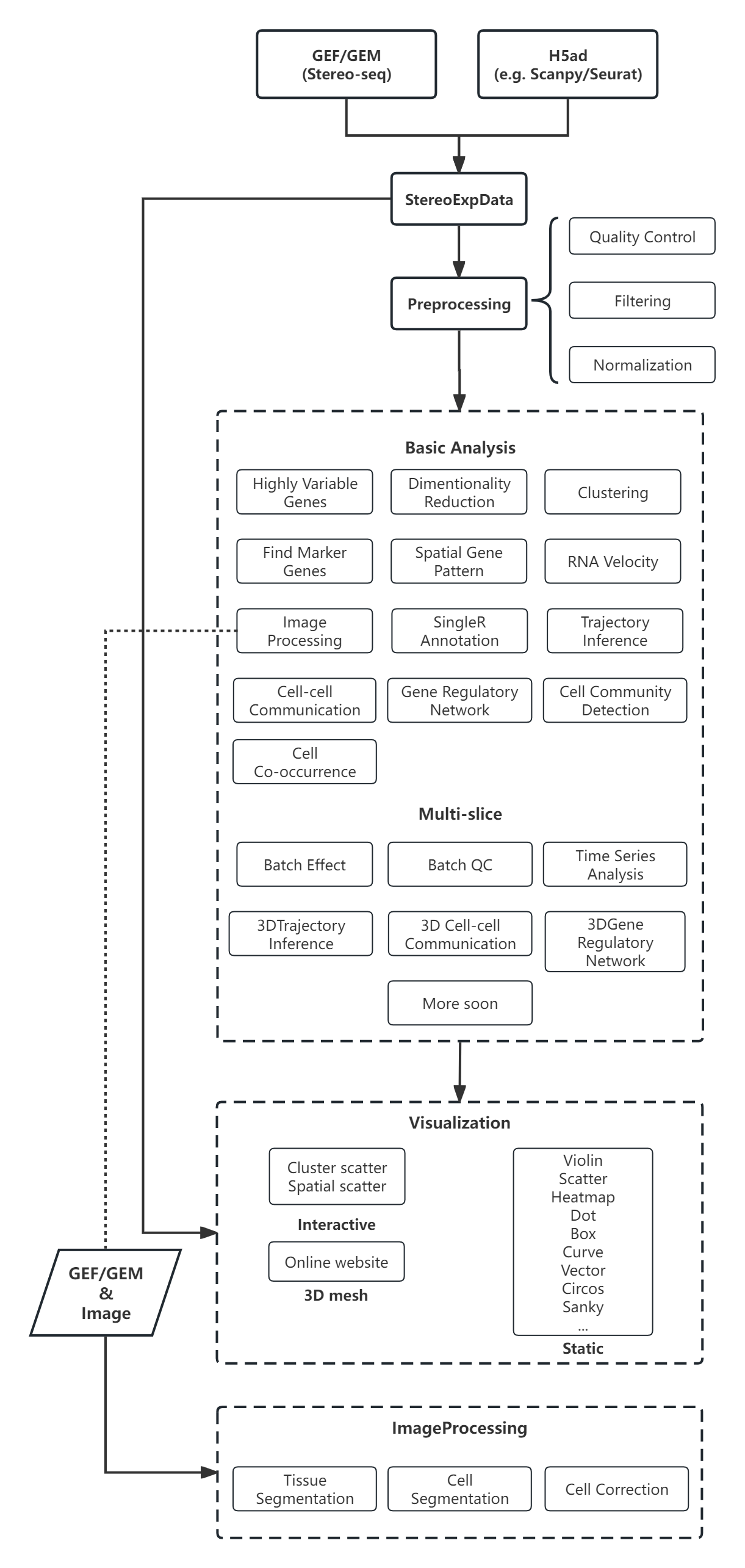
Latest Additions¶
Version 1.3.0¶
1.3.0 : 2024-05-31
Features:
Addition of MSData.tl.st_gears for spatial alignment of Multi-sample.
High Resolution Matrix Export can support both GEF and GEM files.
Addition of parameters
min_countandmax_countfor st.tl.filter_genes.MSData.integrate can be compatible with sparse matrix when
MSData.var_typeisunion.Addition of MSData.tl.set_scope_and_mode to set
scopeandmodeglobally on Multi-sample analysis.Addition of MSData.plt.ms_spatial_scatter to plot spatial scatter plot for each sample in Multi-sample separately.
BUG Fixes:
Fixed the problem that
st.io.read_gemis incompatible with GEM files containing geneID.Fixed the bug of losing part of metadata when writing StereoExpData / MSData into Stereo-h5ad or h5ms file.
Fixed the incompatibility problem with AnnData when performing
st.tl.sctransform.
Version 1.2.0¶
1.2.0 : 2024-03-30
Features:
st.io.read_gemandst.io.read_gefsupport expression matrix files with geneID information.Analysis results of
find_marker_geneswill be saved into the output AnnData h5ad.Upgraded tissue segmentation algorithm.
Addition of
st.tl.adjusted_rand_scoreto calculate the adjusted Rand coefficient between two clusters.Addition of
st.tl.silhouette_scoreto calculate the average silhouette coefficient of a cluster.h5ad2rds.Ris compatible with AnnData version > 0.7.5, to convert from h5ad to rds files.Addition of the clustering category labels to the graph of
st.plt.paga_compare.
BUG Fixes:
Fixed the error of high memory consumption when converting
X.rawinto AnnData.